|
GENODERMATOSES
|
|
|
|
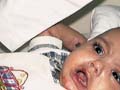
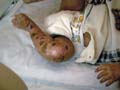
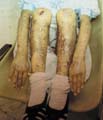
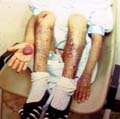
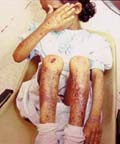
The recessive dystrophic epidermolysis bullosa has the same cutaneous features of the dominant type. Skin manifestations Large, flaccid often-hemorrhagic bullae appear after birth or early in infancy. Mucous membrane manifestations Severe scarring of the mucous membranes of the mouth, pharynx, esophagus and the condition may be fatal. Nail dystrophy is a common complication of the recessive type of epidermolysis bullosa. Secondary bacterial infection is common and causes more complications.
Differential Diagnosis
Treatment Treatment of recessive epidermolysis bullosa is not always curative. High doses of steroids were tried and antibiotic to combat secondary bacterial infection.
This syndrome is an autosomal dominant disease characterized by: Multiple epidermoid cysts . Congenital hypertrophy of the retinal pigmented epithelium Clinical Features Skin manifestations: ‘Sebaceous‘ or epidermoid cysts may appear in early childhood, which may be numerous. These are usually irregularly distributed on the face, scalp, extremities, and are less frequent on the trunk. Cutaneous and skeletal changes may be present without polyposis and polyposis may be present when one or more of the other features of the syndrome is lacking. Lipomas in the subcutaneous tissues, and in other organs, have frequently been noted. Fibromas or desmoid tumors and fibrosarcomas are less frequently present. Gastro-intestinal manifestations: Polyposis of the colon or rectum usually arises during the second decade, but may occur in early childhood. Leiomyomas of the stomach or ileum and malignant changes develop later. Fibromatous growths of the mesentery may be discovered at operation and severe peritoneal scarring may follow surgery. Bone changes Osteomas are usually multiple that develop mainly in the maxilla and sphenoid bones and to a lesser extent in the long bones. Treatment Colonoscopy examinations, where the possibility of prophylactic colectomy should be considered in the second decade.
The ectodermal dysplasia is a genetic heterogeneous group of disorders with a primary defect in hair, teeth, nails or sweat gland function. A. Hypohidrotic Ectodermal Dysplasia
The main features of the syndrome are: Hypohidrosis Hypotrichosis Hypodontia. Clinical Manifestations Reduced sweating. Absent or reduced sweating causes heat intolerance and affected individuals may present with unexplained fever in infancy or childhood. Extreme discomfort can follow exertion or eating hot foods. Teeth manifestations : Total or partial anodontia are the essential features of the syndrome. The temporary and permanent teeth may be entirely absent, or there may be a few teeth present. The incisors and/or canines are characteristically conical and pointed. The jaws develop normally even in complete anodontia but the gums may be atrophic. The conical, pointed teeth are the key feature of the syndrome and may be the only obvious abnormality. In other cases they may be associated with congenital alopecia, and defective sweating, if present, may be detectable only on appropriate testing. Musculo-skletal abnormalities Prominent frontal ridges and chin. Saddle-nose. Sunken cheeks. Thick, everted lips Large ears. Skin manifestations The skin is smooth, soft, and dry, finely wrinkled (especially around the eyes) and appears prematurely aged. Mucous membranes manifestations The mouth may be dry from hypoplasia of the salivary glands and the lacrimal glands may be deficient. Atrophic rhinitis is common and some patients have no sense of smell or taste. Internal organs manifestations Poor development of mucous glands of the respiratory and gastrointestinal tract may increase susceptibility to respiratory infection, dysphagia, stomatitis and diarrhea. Aplasia or hypoplasia of the breasts is occasionally noted. Hair Manifestations Alopecia is often the first feature to attract attention, but it is seldom total. The scalp hair is sparse, dry, and fine and usually remains short. The structure of the shaft may be abnormal. The eyebrows are sparse or absent, but the lashes are usually normal. The beard, pubic and axillary hair is often sparse and other terminal hair on trunk and limbs may be absent. Nail changes The nails are abnormal in about half the cases and may be brittled, thin or ridged, but are seldom grossly deformed. Eye manifestations Ocular abnormalities such as corneal and lenticular opacities may occur. Atopic eczema and asthma are common manifestations. General physical development: may be somewhat stunted, but sexual development is usually normal. Primary hypogonadism is occasionally associated with the syndrome. Mental development: Mental retardation appears in some cases. The expectation of life is normal or only slightly reduced. Treatment Restriction of physical exertion. Choice of suitable occupation. Avoidance of warm climates. Special schooling Psychological support may be needed. Regular dental supervision is essential at an early age. B) Eec Syndrome This is an autosomal dominant syndrome characterized by ectrodactyly (lobster-claw deformity of the hand) with cleft lip and palate. Clinical Manifestations The main features of the EEC syndrome are: Ectrodactyly Sparse wiry and hypopigmented hair Peg-shaped teeth with defective enamel Cleft lip and palate. Lacrimal duct stenosis. Corneal scarring and blindness are serious complications of the syndrome. The normal mucosal covering of the laryngeal folds is absent, and the voice tends to have a breathy quality, as in anhidrotic ectodermal dysplasia. Not all the defects are present in every patient. Sweating may be normal in some patients. C) Rosselli-Gulienetti Syndrome The inheritance of this syndrome is probably determined by an autosomal recessive gene. Clinical Features Hypohidrosis accompanies slight frontal bossing and some depression of the nasal bridge. The scalp hair is often fine, dry, sparse and light in color, the nails are dystrophic and teeth are few and small. Other features are cleft lip and palate, syndactyly and defects of the external genitalia. There may be popliteal web formation. D) Alopecia-onychodysplasia- hypohidrosis Syndrome This is a distinct syndrome of an autosomal recessive inheritance. Clinical Manifestations Hair A child may be born bald and remain so, apart from the few thin yellow hairs on the scalp. Nails : Thick toenails and the fingernails are normal. Hyperkeratosis of the palms and soles knees and elbows and, to some extent, on the skin generally except on the head and neck. Deafness may affect some cases and defects of other organs. Teeth are small. Hypohidrosis.
SYNDROMES ASSOCIATED WITH DNA INSTABILITY Certain genetic syndromes have been grouped together because they share some features such as: Chromosomal instability. Hypersensitivity to irradiation or mutagenic chemicals. The cellular DNA (i.e. the genome) can be damaged by irradiation, or by mutagenic or carcinogenic chemicals in the environment. Normal individuals possess the ability to repair this damage. In some diseases, the ability to repair damage to the genome seems to be impaired. Increased risk of malignancy. These diseases include: Xeroderma pigmontosum, Bloom‘s syndrome, Cockayne‘s syndrome, dyskeratosis congenita, progeria, Fanconi‘s anemia and ataxia telangiectasia.
Xeroderma pigmentosum (XP) is a rare autosomal recessive disease, first described by Kaposi in 1870, characterized mainly by photosensitivity to wavelengths 280 to 310 nm, pigmentary changes, premature skin aging, neoplasia and abnormal defect in DNA repair. Clinical Features Freckles: varying in color and size, appear on the sun exposed areas first on the face and hands and later on other exposed parts, the neck and the lower legs, the lips and the conjunctiva where in severe cases the trunk is affected. The entire face mainly around nose and eyes shows pigmented spots of various tints of brown, mingled with white atrophic patch and telengectasia.
Actinic keratoses, melanomas and squamous cell carcinoma are later manifestations. Telangiectases and angiomas on unexposed skin and on the lingual and buccal mucous membrane have been reported. Small, round or irregular white atrophic spots are common late manifestations. Ocular Lesions The eyes are affected in most cases. Photophobia and conjunctivitis are common early symptoms. Ectropion and destruction of the lower lids expose the bulbar conjunctiva and synblepharon and ulceration may occur. Pigmented macules on the conjunctiva are common. Vascular pterygium, corneal opacities and epitheliomas of the lids, conjunctiva or cornea may develop. Malignant Changes The first malignant tumors may develop as early as the third or fourth year. Basal-cell carcinoma is common and large numbers, sometimes pigmented, may appear over the course of years. Squamous-cell carcinoma is also common. Melanomas arise and may be multiple; they may lead to early death from widespread metastases. The disease is often fatal before the age of 10, and two-thirds die before 20. Neurological Manifestations Neurological abnormalities occur in approximately 20% of XP patients with one or more of the following: mental retardation, areflexia or hyporeflexia, spasticity, ataxia, deafness, dysphasia. De Sanctis-Cacchione syndrome This term has been applied to the association of XP with microcephaly, severe mental deficiency, dwarfism, hypogonadism, deafness, choreoathetosis and ataxia. Post-mortem findings show cerebral and olivopontocerebellar atrophy from neuron loss, without primary damage to white matter, or gliosis. Treatment of Xeroderma General measures Avoid exposure to sunlight. Patients must not go outdoors during daylight hours, except in the early morning or evening, and even then they should wear two layers of clothing and a broad-brimmed hat. All uncovered skin surfaces must be protected by a total sun-block cream, and sunglasses with side shields should be worn. The UVR is harmful up to at least 320 nm, and some fluorescent lights can emit radiation below this wavelength. The relatives of known cases should be carefully examined and tested so those mildly affected individuals may be detected at the earliest possible stage. Treatment of lesions Surgical Early and adequate excision of all tumors is essential and it is to be preferred to radiotherapy on account of the atrophic and degenerate state of the skin. Plastic surgery and grafting of large areas of facial skin may sometimes be required. Topical medications Topical 5-fluorouracil or dermabrasion may be useful for early or premalignant lesions. The eyes may need to be treated with artificial tears, soft contact lenses or even corneal transplant. Systemic medications: oral retinoids may be of help for some cases. Pigmented Xerodermoid The onset of pigmented xerodermoid may be delayed until the third or fourth decade. In pigmented xerodermoid, repair replication is normal, but there is almost total depression of DNA synthesis after exposure to UVR where patients must be protected from sunlight by every possible means. Repair in this type is not impaired but there is almost total depression of DNA synthesis after exposure to UVR. This type appears in older age groups and the manifestations are delayed may up to the third or fourth decade.
POIKILODERMA
CONGENITA Rothmund-Thomson syndrome is a rare autosomal recessive, hereditary disorder, occurring predominantly in females, characterized by cataract and skin degeneration. Clinical Manifestations The skin appears normal at birth. The earliest lesions usually develop between the third and sixth month but sometimes as late as the second year. Skin manifestations Skin lesions begin as tense erythematous and edematous plaques on the cheeks, hand, feet and buttocks followed by fine or punctate reticulated atrophy, telangiectasia, pigmentation, depigmentation and ultimately, the lesions closely resemble chronic radiodermatitis. Hair manifestations Scalp hair is often sparse, fine, and may be absent. Eyebrows and lashes, pubic and axillary hair are often sparse or absent. Teeth changes Teeth are often normal, but microdontia and early caries have been reported. Nails are normal, or small and dystrophic. Photosensitivity on sun exposed areas, which may be severe eliciting bullous reaction. Ocular manifestations Bilateral cataracts, usually between the fourth and seventh year. Neurological manifestations Some cases show mental retardation, while physical development is frequently retarded and the affected patients are dwarf. Endocrine changes: hypogonadism and hyperparathyroidism. Mental development is usually normal, but mental retardation may develop in some cases. Expectation of life appears to be normal. Diagnosis Age of onset: is one of the essential features in differential diagnosis. Distribution of skin lesions mainly on the sun exposed areas. Skin lesions are characterized by atrophy, telangiectasia and mottled pigmentation, most intense on light-exposed skin. Photosensivity is a feature of the syndrome. Differential Diagnosis
Treatment of Poikeloderma Protection against sunlight is important and early diagnosis of any malignancy. Retinoids may help some cases.
The inheritance of this rare syndrome is determined by an autosomal recessive gene. Clinical Features Cerebellar ataxia is apparent as soon as the child begins to walk. Dysarthria. Mental and physical development is retarded. Congenital cataracts associated with rotary and horizontal nystagmus. Skeletal defects are commonly present. The teeth are malformed and the lateral incisors may be absent. The nails are thin and fragile. The hair is sparse, fine, short, fair and brittle.
SECKEL‘S
SYNDROME The syndrome is a rare inherited by an autosomal recessive gene. Clinical Features Main features: Prominent beak-like nose Growth retardation, microcephaly and mental deficiency. Other manifestations: Hypoplastic face with large eyes. Skeletal defects are frequent. The hair may be sparse and prematurely gray. Diagnosis The syndrome must be differentiated from others in which there is intrauterine dwarfism.
CARTILAGE-HAIR
HYPOPLASIA SYNDROME This is a rare syndrome, characterized by high degree of dwarfism and associated with multiple skeletal deformities resulting from a metaphyseal dysostosis. Clinical Features Hair changes The hair is sparse, short, of very fine caliber, lighter in color than in unaffected siblings and is often very brittle. Some affected individuals are almost bald. Sexual development is normal. Immunological abnormalities. Increased susceptibility to infections. Decreased number of circulating T- and B-lymphocytes. Metaphyseal chondrodysplasia occurs in association with immunodeficiency, but the hair is normal.
This is an autosomal dominant syndrome characterized by: Lower-lip pits Lip pits, the most common, which vary from mere depressions to deep channels up to 15 mm, may secrete small quantities of viscous saliva. Cleft lip and/or palate Hypodontia. Syndactyly is sometimes associated with the syndrome.
This rare syndrome affects infants and is usually present from birth. Clinical Manifestations Progressive arthropathy. Urticarial rash Uveitis and mental retardation. Central nervous system involvement and deafness may occur. Bone deformities. Epiphyseal abnormalities, periosteal elevation along the shafts of the long bones. Short stature, delayed fontanel closure, frontal bossing and a broad nasal bridge.
Clinical Feature The main manifestations of this syndrome are: Alopecia Nail dystrophy Ophthalmic complications. Thyroid dysfunction Hypohidrosis Ephelides Respiratory-tract infections Enteropathy
|

| Contents | << Previous Chapter | Next Chapter >> | Search |