|
SKIN MANIFESTATIONS OF METABOLIC DISEASES
|
|
|
|
Porphyrins are metabolic by-products, that have not followed the usual synthesis from glycine and succinyl co-enzyme A to heme with production of Porphobilinogen and aminolevulinic acid. Different factors such as drugs , chemicals and hormones can increase porphyrin synthesis. The site of disturbance is either in the liver (hepatic porphyria) or in the bone marrow in the erythroid cells (erythropietic porphyria ). Types of porphyria
The clinical manifestations are due to abnormal Porphyrins metabolism. Drugs such as barbiturates, sulfonamides, chloramphenicol, chloroquine, griseofulvin and toxins, fungicides (hexachlorobenzene), may cause this type. Different types of porphyria can cause different skin , hair and nail manifestations: Clinical Manifestations Skin manifestations The skin is fragile, tear easily and forming blisters due to dermo epidermal separation. Poikeloderma like reaction in the form of pigmentation , atrophy and telengectasia on sun exposed areas. Hypopigmentation and scarring after healing. Hypertrichosis is not a common manifestation of porphyria cutanea tarda. Photosensitivity The patients are sensitive to sunlight even when they are indoors. Patients are labile to have phototoxic reactions. Associated diseases Liver cirrhosis, hemochromatosis, carcinomas and Hodgkin‘s disease may be associated with porphyria cutanea tarda. Diagnosis Urine shows a pinkish coral red fluorescence under Wood‘s light . Positive bromosulphalin test. Detection of Porphyrins in urine and feces . Three-step procedure (devised by Castro): Disposable plastic column charged with anion exchange resin, permits detection of various porphyrins as well as their precursors. Hereditary Porphyria Cutanea Tarda This is a rare type, which is carried as a dominant gene and appears at early age around 15 years of age. Treatment Phlebotomy: 500ml of blood every two weeks. Usually 3000-5000 ml of blood is taken . Involution of skin lesions usually appear after the second blood intake . Chloroquine: 500mg twice weekly for the adult age are believed to have an encouraging results. Sodium bicarbonate: used to alkalinize urine may have a beneficial effect.
ACUTE
INTERMITTENT PORPHYRIA This type is characterized by periodic attacks of abdominal colic, gastrointestinal disturbances, paralysis and psychiatric disturbances. Clinical Manifestations: General manifestations: Abdominal colic. Peripheral neuropathy. Psychiatric abnormality. Skin manifestations: Skin pigmentation. Hirsutism. Photosensitivity is not a feature of this type .
Skin manifestations include those of porphyria cutanea tarda and acute intermittent porphyria but occurring in earlier age groups.
Photosensitivity causes polymorphous light eruption leading to pruritic, erythematous papulo vesicular and urticarial rashes mainly on the sun-exposed areas. Skin lesions heal leaving linear pitted scars .Other manifestations are purpura , oedema and severe burning pain.
This type appears early in childhood from 2-5 years of age and inherited as a dominant trait. Photosensitization is a characteristic feature of erythropoietic protoporphyria . In this type it is believed to be due to the longer wavelengths of ultraviolet (UVL) which ranges from 320-450nm. Ordinary window glass offers no protection from the effect of sun on such patients. Clinical Manifestations Skin lesions are pruritic erythematous, plaque-like edema, wheals and even vesicles or bullae on the sun exposed areas. The skin lesions may heal with scarring with waxy thickening of the nose, cheeks, over the proximal finger joints, circumoral atrophy and scarring. Diagnosis Characteristic cutaneous lesions. Photosensitivity. Increased proto-and coproporphyrins in feces. Increased porphyrins in red blood cells . Fluorescent microscopic examination of blood : Few drops of blood are diluted 1:5 with normal saline are placed on a microscopic slide and examined by the oil immersion objective of a fluorescent microscope. Erythrocytes usually show characteristic fluorescence. It should be noted that in this type of porphyria , urine usually does not give fluorescence under the Wood‘s light .
CONGENITAL ERYTHROPOIETIC PORPHYRIA This type is a hereditary disease, transmitted by an autosomal recessive gene. Clinical Manifestations Skin manifestations Skin manifestations appear early in infancy on the sun-exposed areas that are due to photosensitivity. Painful bullous lesions, which heal by destructive and disfiguring scarring and causing destruction of the cartilage of the nose, ears, and nails. Cicatricial alopecia Hypertrichosis: with hair on the cheeks, profuse eyebrows, and long eyelashes (“monkey face“). Urine shows high amount of copro and uropophyrines. Diagnosis Congenital erythropietic porphyria has the following characteristics that are diagnostic even in early infancy: Red urine in early infancy . Photosensitivity Hemolytic anemia Splenomegaly Erythrodontia of both deciduous and permanent teeth. Coral red fluorescence of the teeth when exposed to Wood‘s light.
This disease affects children with blond hair, blue eyes and fair skin due to lack of the enzyme phenylalanine hydroxylase, which is essential for degradation of phenylalanine to tyrosine. Clinical Features: Photosensitivity. Eczema like reaction. Secondary infections are common. Scleroderma-like skin lesions. Induration of thighs and buttocks are common manifestations in affected infants and children. General manifestations. Mental deficiency. Epileptic seizers. Laboratory findings. Presence of phenyl pyruvic acid in urine. This can be easily detected by adding to urine few drops of ferric chloride solution that will give deep green color. Treatment Special diet for infants and young children containing low phenylalanine and this should be given immediately after birth.
This is a hereditary disease transmitted as an autosomal recessive gene, due to enzymatic defect in the metabolism of tyrosine and phenylalanin. Clinical Manifestations. Dark urine, which becomes later black due to increased homogentisic acid secretion in urine. Deposition of brown-black pigment in the connective tissue. In older age groups the manifestations are: Pigmentation of the sclera, which is an early sign. Deposition of pigment in the cartilage of the ears, nose and tendons of the extremities which may show blue, mottled brown macules. Internal organs mainly great vessels, valves and larynx, genitalia may be also involved . Arthropathy affecting the spinal joints , hips ,knees and shoulders.
ABNORMAL
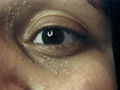
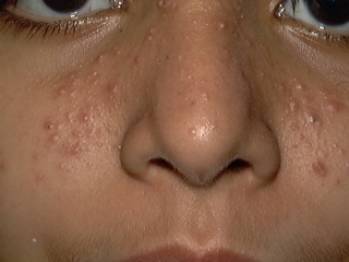
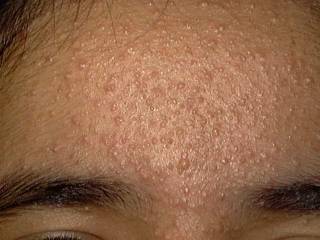
LIPID METABOLISM Xanthomatosis is accumulation of lipids in association of foam cells in the tissues. Different clinical types. Xanthelasma palpebrarum: this is the most common type of xanthomas affecting any age. Middle age women are the commonest to have this problem especially those who have biliary diseases. The lesions are yellowish plaques on the eyelids, which may coalesce to form large plaques. Plane xanthomas: yellowish, raised papules, symmetrically distributed mainly on the eyelids sides of the neck and palms. Eruptive xanthomas: yellow papules appear on the extensor surfaces of the limbs, joints and buttocks surrounded by a rim of erythema and may be tender. Eruptive xanthoma is associated by increased serum triglycerides .
Tendinous xanthomas: nodular yellowish lesions appear on the tendons on the extensor surface due to cholesterol infiltration. Tuberous xanthomas: symmetrical nodular lesions, appear over the extensor surface of the joints and accompanied by increased serum triglycerides and cholesterol.
MANIFESTATIONS OF HYPERLIPOPROTEINEMIA
Generalized xanthelasma Histiocyhtosis X Litterer -Siwe disease Hand -Schuller-Christian disease. Eosinophilic granuloma. Juvenile xanthogranuloma. Refsum‘s Syndrome This syndrome is a genetic disorder of lipid metabolism. Neurological and cutaneous features characterize this syndrome. The underlying abnormality is a deficiency in phytanic acid, displacing unsaturated fatty acids as linolenic acid from tissue lipids. Clinical Manifestations Skin manifestations are mainly dryness of the skin, which simulate icthyosis vulgaris. General manifestations begin early in childhood similar to retinitis pigmentosa with different neurological (polyneuropathy), ataxia, cardiac and bone manifestations. Various neurological changes occur, including deafness, cerebellar degeneration, polyneuropathy, and retinitis pigmentosa and cardiac abnormalities. Refsum‘s syndrome can be diagnosed by lipid analysis of the blood or the skin. Normally no or very little phytanic acid is found in the blood (0-33 mumol/l). Treatment Treatment by a phytanic acid-free diet, in which green vegetables and dairy products are excluded, has been used. Plasma exchange in conjunction with diet.
ABNORMAL AMINO - ACIDS METABOLISM These changes are recessively transmitted error of metabolism of amino acids leading to different skin manifestations. The skin manifestations depend on the specific amino-acid metabolism abnormality.
This syndrome is due to deficiency in homogentisic acid oxidase leading to accumulation of homogentisic acid that can be detected in urine. Clinical Features In early life: dark urine and sweat. Arthropathy of spines and knees due to chondreal cartilage thickening. In older age groups the skin of forehead, ears, cheeks and around the eyes is pigmented. Pigmentation of the sclera.
This disease is due to disorder in methionin metabolism due to an absence of hepatic cystathione synthetase causing abnormality in collagen formation. Clinical Features The clinical manifestations appear early, in the first year of life. Thin, yellowish skin and atrophic scars. Fine sparse hair, which brittles easily due to disulfide bond reduction. Intravascular clotting leading to livedo reticularis. Treatment Diet low in methionin. Supplement by pyridoxine and cysteine may give good improvement.
This error of metabolism of tryptophane leads to nicotinic acid deficiency. The changes occur early in infancy due to unabsorbed tryptophene, which is broken into the gut to indoles that is absorbed, metabolized and excreted in urine as indican. Clinical Features Skin manifestations are like pellagra in the form of dry, scaly and sharply demarcated rash on the sun-exposed areas. Photosensitivity. Neurological manifestations: ataxia and mental retardation. Diagnosis By the clinical picture. Pellagra like eruption. Neurological manifestations, ataxia and mental disturbances. Urine examination shows increase in indican and monocarboxylic amino acid. Treatment Nicotinamide. Treatment of skin manifestations by emollients and keratolytics as topical salicylic acid in an ointment base alone or in combination with steroids (Locasalene). Sunscreens and avoiding too much exposure to sunlight.
These metabolic disorders are due to a defect in specific enzymes leading to accumulation of intermediary metabolic products in lysosomal organelles. These syndromes include Hurler‘s syndrome, Chediak-Higash syndrome and others.
This disease appears early in infancy in the first months of life. Clinical Features Skin manifestations The skin manifestations are brown, scaly papules on the seborrheic areas on the scalp, behind the ears, naso-labial folds and mid chest. Systemic manifestations include purpura, systemic histiocytosis and malignancy. Most infants die in the first two years from infections mainly due to pneumonia. Treatment Treatment is not always curative. Antibiotics for pulmonary infections. Corticosteroids. Cytosine and blood transfusion can be tried.
ANDERSON‘S
FABRY DISEASE This is a rare X linked recessive trait storage disorder leading to accumulation of ceramide trihexose in the tissues mainly in the endothelium of smooth muscles and blood vessels. Clinical Manifestations This syndrome has complex skin and systemic manifestations. Skin manifestations begin to appear in the adult age in the form of dark blue or black lesions mainly on the back, abdomen, buttocks, umbilicus and mouth. Systemic manifestations appear early in childhood in the form of weakness, malaise, cramps. In adult age, there is vague symptoms as fever after exercise with decreased sweating, neurological and psychological episodes and severe pain of the feet and hands. Serious and even fatal complications in older age groups are due to cardiac, renal and cerebrospinal accidents.
Both of these syndromes are genetic diseases due to error of metabolism of mucopolysaccharides leading to greatly thickening of the skin due to deposit of mucopolysaccharides in tissues limiting joint movements.
|
| Contents | << Previous Chapter | Next Chapter >> | Search |