|
CUTANEOUS
VASCULAR DISEASES
|
|
|
|
Petechiae are small, purpuric lesions up to 2 mm across while ecchymoses or bruises are larger extravasations of blood. Purpura is normally distinguished from erythema when pressure is applied by finger or by pressure of a slide on the erythematous patch (diascopy )fails to blanch the lesion. The characteristic color changes in purpuric lesions vary from purple, orange, brown and even blue and green. Discoloration of the skin or mucous membrane is due to extravasations of blood. Types of Purpura Classification of purpura is usually unsatisfactory. There are different classifications in the different textbooks depending either on the morphological or etiological characters of these diseases.
Etiology Thrombocytopenia purpura may be primary (idiopathic ) due to unknown causative factors or secondary to different agents.
Clinical Manifestations Thrombocytopenic purpura may occur at any age, but in two-thirds of cases it occurs in young age . Females are more commonly affected than males. The onset may be gradual or, acute especially in children. There is an appreciable mortality, especially in the acute form, mainly from cerebrovascular accidents. Bleeding occurs into the skin with areas of petechiae or larger hemorrhages and may occur in internal organs. Joint involvement is unusual. The spleen may be slightly enlarged . Spontaneous remission occurs in a proportion of acute cases, but is rare in chronic cases of more than 3 months‘ duration in which a continuous or fluctuating course may occur. Diagnosis Clinical picture. Blood picture: low platelet count , Megakaryocytes are present in normal or increased numbers in the bone marrow. Negative bone-marrow findings. Differential Diagnosis Systemic lupus Drug-induced purpura Disseminated intravascular coagulation Renal failure.
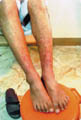
Vascular purpura Non-thrombocytopenic purpura comprises the vast majority of cases of purpura. Blood may leak as a result of: Damage to small blood vessels. Increase in the intraluminar pressure. Deficient vascular support. Bleeding may arise from a disturbance of one or more of the following mechanisms:
Etiology of Vascular Purpura.
Purpura may be the presenting and sometimes the only symptom of disturbances in plasma proteins. It may occur with cryoproteinaemia. This type appears most commonly on the unprotected parts after exposure to cold. Hyper globulinaemia due to different causes such as idiopathic (Waldenstrom‘s) sarcoid, lupus erythematosus, Sjogren‘s syndrome, myeloma, may give rise to purpura. The clinical features of dysproteinaemic purpura are erythematous papules that occur mainly on the legs and rapidly progress to form punctate purpuric lesions In mild cases the eruption disappears within few days, but in more severe cases the purpura becomes confluent and permanent. A similar pattern has been reported in association with cystic fibrosis, whether or not associated with cryoglobulinaemia.
HENOCH-SCHOENLEINE
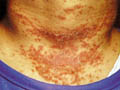
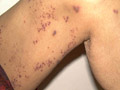
PURPURA This type affects children and young adults . Urticaria and purpura with multisystmes involvement of kidneys, bowel and joints characterize this type of purpura. Etiology Damage to the walls of small blood vessels due to deposition of immune-complex substances. Cryoglobulins have been found rather than the immune complexes. An antigen associated with upper respiratory tract infection is suspected to be part of the usual cause of the immune response. Clinical Manifestations General manifestations: The manifestations usually begin with mild fever, sore throat, and upper respiratory tract infections which may precede the skin rash. Skin manifestations: Macular rash appears first on the extensor surfaces of the limbs and buttocks, which becomes rapidly, urticarial and purpuric with central necrosis of the lesions. Systemic manifestations Renal involvement, which is focal nephritis. This is a serious manifestation of the disease. Bowel involvement leads to abdominal colic and hemorrhage. Polyarthritis and pain in the joints are another manifestation. The course of the disease is chronic . It may take weeks for regression of the skin lesions, but usually there is recurrence. Renal and bowel manifestations may improve or may cause serious complications.
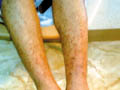
These are vascular diseases of undetermined cause with different manifestations and share the same histopathological features. These include different diseases mainly: Schamberger‘s Disease This is a progressive pigmented purpuric dermatosis of unknown etiology, affecting male children and other age groups that may show familial incidence. Clinical Manifestations The skin lesion is irregular brown plaques that may present with different pigmentations due to hemosidrin deposits. ‘Cayenne pepper‘ spots characterize the lesions. The condition is usually asymptomatic , although there may be some slight itching. The eruption is characteristically very chronic and may persist for many years. The pattern of the eruption changes where these may show slow extension with some clearing of the original lesions. Spontaneous cure may occur eventually. Differential Diagnosis Drug eruption: different types of drugs particularly carbromal and other drugs may cause similar types of purpuric skin lesions. Food allergy and food additives. Clothing dermatitis. Hyperglobulinaemic purpura. Early mycosis fungoides .
ITCHING
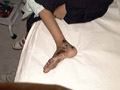
PURPURATOUS Itching purpura is an eczema like purpura, which begins usually as an eczematous purpuric reaction around the ankles and spreads peripherally. The eruption often has a rather characteristic orange color. Eczematous skin lesions presenting as erythematous purpuric macules that may simulate shoe dermatitis or drug reaction. The condition rarely becomes generalized, affecting mainly exposed areas due to chaffing or friction. Spontaneous improvement is usual, but recurrences may occur. Differential Diagnosis Drug reactions:Carbromal sensitivity, Meprobamate and Carbamazepine. Food allergy . Clothing or rubber contact dermatitis. Schamberg‘s disease is distinguished by its more persistent course and by the usual lack of itching.
LICHEN
AUREUS This is a more localized, more intensely purpuric eruption. Clinical Manifestations Skin lesions begin as rust-colored to purple non-itchy solitary macules, seldom truly golden, which may resemble a bruise. Small vesicles may be seen in its course of the disease, that may persist for few years. Histopathological changes are in the form of capillaritis, infiltration with lymphocytes and histocytes. Treatment Topical steroids may be helpful.
PURPURA
ANNULARIS TELENGECTODES This type of capilliritis may show familial tendency that affects mainly young adults of both sexes, where any age is not immune. Clinical Manifestations Lesions occur at any site, often in the absence of venous stasis and may be few in number or very numerous. Skin eruption presents with small annular plaques, telangiectasia and haemosidrin deposits causing purple, yellow or brown patches that may contain ‘cayenne pepper‘ spots. Individual lesions persist unchanged for many months or years, or there may be slow centrifugal extension. Sometimes the lesions disappear and may recur with the same eruption. Treatment The lesions are asymptomatic and rarely treatment is needed .
These diseases are due to defects in one or more of the numerous factors related to clotting with abnormalities of the platelet functions .
Vascular purpura is uncommon in the neonatal period but may occur. Hemorrhagic disease of the newborn is due to an accentuation of the normal fall of prothrombin within the first week of life. Differential Diagnosis Purpura or bleeding within the first month of life should be differentiated from different types of purpuric skin diseases: Deficiency of the clotting factors . Deficiency of the protein S or protein C. Hemophilia and other bleeding diseases, which rarely cause bleeding at this age. Thrombocytopenia may be due to congenital failure of megakaryocytes. Immunological mechanism in a child whose mother has idiopathic thrombocytopenic purpura or systemic lupus erythomatosus. Neonatal rubella and Wiskott-Aldrich syndrome. Haemangiomas.
Cutaneous systemic angitis is a complex and widespread necrosis of the small blood vessels. Etiology Different factors are blamed to be the cause of systemic angitis. These include: Drugs: the most common drugs which can cause systemic angitis are :sulfonamides, acetylsalicylic acid (Aspirin), phenothiazines, barbiturates. Infections: Streptococcal infection , pyodermas , upper respiratory tract infection . Insecticides and weed killers. Cutaneous systemic angitis includes different diseases mainly :
Purpura fulminans is a serious disorder affects patients of different ages, but most commons in children. Clinical Features Lesions are characterized by the development of more or less symmetrical and well-defined “lakes“ of confluent ecchymosis without petechiae mainly on limbs, trunk and face. The onset is sudden, and the lesions enlarge rapidly, with coalescence and often with the development of hemorrhagic bullae and central necrosis. There is a surrounding erythema and the lesions are tender. The patient is frequently febrile. Vascular thrombosis is a particular feature of this disorder. There is a substantial danger of internal hemorrhage, shock and death. Etiology In older children, purpura fulminans may have several causes. It is a highly characteristic feature of meningococcal septicemia and may occur as a sequel to a number of other infections, including common infections such as streptococcal infections, varicella, and measles. In the neonate, however, its occurrence is very suggestive of protein C deficiency . Treatment Treatment comprises rapid transfusion of fresh frozen plasma.
INFANTILE ACUTE HEMORRHAGIC EDEMA It is a distinctive disorder, comprising a combination of purpura, often in a cockade pattern, and an inflammatory edema of the limbs and face, occurring almost exclusively in children under the age of 2 years, with a tendency to recurrence in the short term and subsequent spontaneous resolution . The cause of infantile hemorrhagic edema remains unknown, though it may represent an infantile analogue of Henoch-Schonlein purpura.
DISSEMINATED
INTRAVASCULAR COAGULATION Disseminated intravascular coagulation may produce a clinical picture varying from a severe and rapidly fatal disorder to a relatively minor disorder. Predisposing Factors This is due to congenital or an acquired deficiency of the protein S and protein C components of the anticoagulation system. Etiology The causes of disseminated vascular coagulation are: Extensive tissue damage . Severe infections (especially Gram negative septicemia). Immune reactions. Malignant disease. Snake bites. Giant haemangiomas. The normal inhibitory mechanisms of clotting are over-coming, so that there is intravenous coagulation, followed by consumption and depletion of platelets and plasma clotting factors. Clinical Manifestations The manifestations include bleeding, thrombo-embolism and hemolytic anemia. The onset may be acute, sub acute or chronic. Mild cases: show petechiae, purpuric papules, hemorrhagic bullae and acral cyanosis. There is decreased fibrinogen and increased fibrin degradation products. Skin biopsy may be useful in showing intravascular thrombi. Severe cases: the onset is sudden, with high fever and a very extensive, usually symmetrical, purpuric rash of the extremities. A fatal outcome may follow within 2 or 3 days. Treatment Treatment of shock and replacement therapy with platelets, fibrinogen, and fresh frozen plasma. Symptomatic treatment. Treatment of the cause. The role of heparin is still somewhat controversial.
MANIFESTATIONS OF CUTANEOUS SYSTEMIC ANGITIS Skin manifestations The lesion usually begins in the lower legs, buttocks , hands and wrists. Mucous membrane lesions are rare. Different skin lesions may appear either purpuric rash, hemorrhagic vesicles and bullae may develop. Finally there are nodules and ulceration, which persist for a long period .Usually one type of these lesions, manifest either purpuic rash alone or vesicular type. General manifestations Fever, malaise and myalgia are frequent symptoms . Burning sensation and pain may be mild or sometimes severe depending on the site and extent of the lesions. Arthralgia and swollen joints. Kidney involvement : leads to manifestations of glomerulonephritis. Gastrointestinal manifestations: haematemesis, melena, peptic ulceration, esophageal ulceration. These usually manifest with nausea , vomiting , diarrhea and anorexia . Congestive heart failure manifestations. Lung involvement leads to pneumonitis. Eye changes: retinal hemorrhage. Neural manifestations: peripheral neuritis , diplopia , dysphagia and hoarseness of the voice. Diagnosis Laboratory Tests: ESR: is usually elevated Hyperglobulinaemia. IgG globulin and C3 complement appear in the areas of fibrinoid necrosis of the blood vessels . Treatment Treatment of the cause. Corticosteroids may help some cases.
Cutaneous vasculitis is characterized by purpuric or necrotic urticarial lesions and may be associated with vasculitis of internal organs. Histopathological changes of cutaneous vasculitis are characteristic. These include fibrinoid changes in the small dermal blood vessels with polymorphonuclear and ‘nuclear dust infiltrate. Blood Picture. The ESR may be normal, but is usually raised. When ESR is much raised in urticarial vasculitis, lupus erythematosus should be excluded. Neutrophilia or eosinophilia may occur. Hypocomplementaemia is usual. Circulating immune complexes are often demonstrated . Differential Diagnosis Infection, drug intake, internal neoplasia or collagen disease, where these may show cutaneous vasculitis and should be excluded .
|
| Contents | << Previous Chapter | Next Chapter >> | Search |